P. aeruginosa PA14 Genomic Sequencing Project
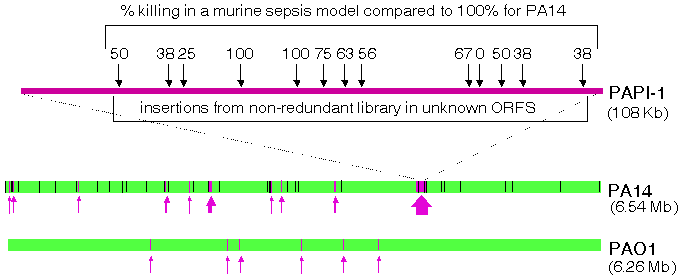
PA14 Sequencing and Comparative AlignmentsOur PGA determined the complete sequence of the PA14 genome sequence collaboratively with the Harvard Partners Center for Genetics and Genomics (HPCGG, Cambridge, MA). The primary rationale for obtaining the PA14 sequence was to facilitate the construction of the non-redundant PA14 transposon mutant library because there is a significant amount of PA14-specific sequence that is not present in the genome of the publicly available sequence of P. aeruginosa strain PAO1. PA14 Genomic DNA was subjected to double-ended shotgun sequencing and the resulting sequence was analyzed using Phred, Phrap, and Consed . The shotgun phase of the sequencing project has been completed, yielding 6.54 Mb of PA14 sequence (10.2-fold coverage), compared to 6.26 Mb for PAO1. The current assembly consists of a single large contig. Only one gap remains, which is thought to be less than 3.5 kb. This sequence has been deposited in GenBank (accession number AABQ00000000) and can be downloaded here. Alignments of PAO1 with the current PA14 assembly are being carried out using a combination of BLAST searches and analysis with MUMmer, a tool for global alignment of genomes. Our preliminary comparisons of the two genomes (Figure 1) have demonstrated that they are extremely similar but have also pinpointed regions of differences, including a 107911bp insertion in PA14 that is absent in PAO1 (P. aeruginosa Pathogenicity Island 1, or PAPI-1). Approximately 96.3% of the DNA sequence found in PAO1 is also found in PA14 and approximately 92.4% of PA14 DNA sequence is found in PAO1. Our PGA, in collaboration with the Rahme lab at MGH, identified and characterized two novel P. aeruginosa pathogenicity islands (PAPI-1 and PAPI-2) in the genome of PA14, which are absent from the reference strain PAO1 (He et. al, 2004, PNAS 101:2530-2535). The 108 kb PAPI-1 and 11 kb PAPI-2 exhibit highly modular structures and contain many of the 313 PA14-specific ORFs. More than 80% of the PAPI-1 DNA sequence is unique, and 75 out of the 115 predicted ORF products are unrelated to any known proteins or functional domains. PAPI-2 harbors 15 predicted ORFs and 8 of these are absent from strain PAO1. On the other hand, most of the genes within these islands that are homologous to known genes occur in other human and plant bacterial pathogens. Importantly, many PAPI-1 ORFs are also found in several P. aeruginosa cystic fibrosis isolates.
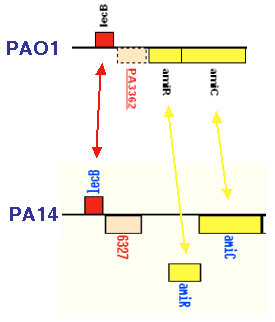
We have generated and analyzed 23 mutant strains, including 10 non-polar deletions and 13 non-redundant library mutants, to assess whether the PAPI-1 and PAPI-2 ORFs that encode hypothetical/unknown functions promote P. aeruginosa pathogenesis. This work showed that both islands carry genes that allow PA14 to thrive on evolutionary diverse hosts, including plants (Arabidopsis) and mammals (mouse). 20 out of the 23 ORFs mutated, encode functions necessary for plant or mammalian virulence, and 12 are required for "wild-type" virulence in both hosts (Figure 1 shows the locations and phenotypes of the mutants obtained from the non-redundant library). Although the majority of these genes encode products of unknown function, their presence in P. aeruginosa clinical isolates, including those from CF patients, suggests that they encode conserved functions important for fitness and survival. PA14 Genome Annotation and Proteomic AnalysisCharacterization of novel pathogenicity factors such as those described above should provide insights into broad host pathogenic and defense mechanisms. Importantly, completion of the PA14 genome sequence annotation, currently in progress, might also lead to the identification of additional PAPI blocks and novel virulence genes in the remaining PA14-specific ORFS that have not yet been studied. The annotation of PA14 was initiated with an automated ORF prediction phase utilizing both BLAST and Glimmer software tools and this list of putative ORFs is currently being manually edited as a collaborative effort involving researchers from MGH and Harvard Medical School (Boston MA). A draft version of the PA14 annotation will be posted on this site when it is completed. To facilitate the manual annotation effort, we have initiated a proteomic analysis of PA14 in collaboration with the MS Proteomics Center at the Harvard Partners Center for Genetics and Genomics (HPCGG, Cambridge, MA) and with the laboratory of George Church at Harvard Medical School (Boston, MA). Briefly, PA14 are grown in liquid media, harvested in mid-log phase, and sonicated in the presence of Urea and detergents. The complex protein mixture is reduced and alkylated and subjected to SDS-PAGE to separate it into several molecular weight fractions. Tryptic digests are performed in-gel, and the resulting peptides are analyzed by mass spectrometry. In a pilot analysis, we have positively identified proteins that have previously been experimentally verified in PAO1 (or other P. aeruginosa strains), proteins corresponding to ORFs annotated in PAO1 as hypothetical an not experimentally validated, as well as hypothetical ORFs in PA14 that do not appear in PAO1. An example of the latter case is shown in Figure 2. We are currently increasing the scale of the proteomic analysis to sample the entire PA14 proteome.
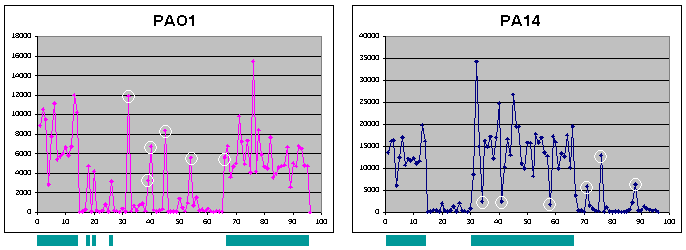
Genotyping of P. aeruginosa Strains Using MicroarraysRegions of PA14 absent in PAO1 but conserved in other clinical isolates of P. aeruginosa are of particular interest to our group. To determine, on a genome-wide level, whether "PA14-specific" sequences (i.e. - absent in PAO1) are conserved in other strains, we are developing a P. aeruginosa spotted oligo microarray in collaboration with the MGH Microarray Core. By designing probes for sequences present in PA14 and absent in PAO1, or vice versa, a large number of unsequenced P. aeruginosa strains can be analyzed by labeling their genomic DNA and hybridizing to the array to determine which sequences appear to be conserved in virulent strains. The initial version of this array will focus primarily on sequences that are present in PA14 but absent in PAO1, present in PAO1 but absent in PA14, as well as additional sequences found in other P. aeruginosa strains but absent in at least one of the sequenced strains (suggestions for additional P. aeruginosa sequences to be included in later versions of the microarray are welcomed). Controls include sequences that are found in both PA14 and PAO1 and likely to be present in all P. aeruginosa strains (e.g. - essential housekeeping genes) as well as sequences likely to be absent in all P. aeruginosa strains. The next generation of this array will be a full genome chip, including probes for all PA14/PAO1 sequences as well as sequences from other strains. This chip will be made available to the research community and can be utilized for genotyping of strains as described above, for traditional transcriptional profiling studies, as well as being used in conjunction with the PA14 mutant library for high-throughput screens of pooled mutants utilizing a methodology called TraSH (Transposon Site Hybridization). A pilot microarray of 95 oligos (and one blank) spotted in quadruplicate has been produced to validate its utility for genomic typing. Included in the microarray are 14 sequences present in both PA14 and PAO1, 36 sequences found in PA14, 32 sequences found in PAO1, and 13 (P. aeruginosa) sequences found in neither strain. PA14 and PAO1 genomic DNA was fragmented with a restriction enzyme (Hae III), labeled with cy3 or cy5, and hybridized to the array (Figure 3). Since the two labeled samples yielded different levels of overall hybridization, each data set was normalized (by dividing each hybridization intensity by the average hybridization intensity for the total sample) and an arbitrary cut-off was determined: a normalized intensity of >0.5 resulted in the sequence being assigned as present, and a normalized intensity of <0.5 was used to assign sequences as absent. Using these criteria, 94% of the probes were correctly assigned as present or absent in each of the two strains. We are currently examining the specific probes that have led to incorrect assignments in either or both of these strains and to manipulate the hybridization conditions to improve accuracy with the two sequenced strains. The first generation (non-pilot) array containing 465 probes (PA14-specific, PAO1-specific, or control sequences) is scheduled for production and an initial panel of 22 strains (including lab strains, environmental isolates, and clinical isolates from a variety of infection sites in the human body) will be analyzed. Finally, a second generation array that comprises a (near-)complete set of all P. aeruginosa sequences will be designed for genomic typing and transcriptional profiling experiments as well as additional sequences custom designed for our PA14 mutant library to allow for analysis of pooled mutants utilizing TraSH.
| ||||||||||||||