PA14 Transposon Insertion Mutant Libraries
I. Description of the PA14 non-redundant transposon insertion libraryComprehensive screening for P. aeruginosa virulence factors requires a library of P. aeruginosa mutants containing a disruption in each gene comprising the bacterial genome. We are constructing a non-redundant library of PA14 transposon insertion mutants such that each non-essential gene in the P. aeruginosa genome will be represented by one transposon insertion mutant in the library.
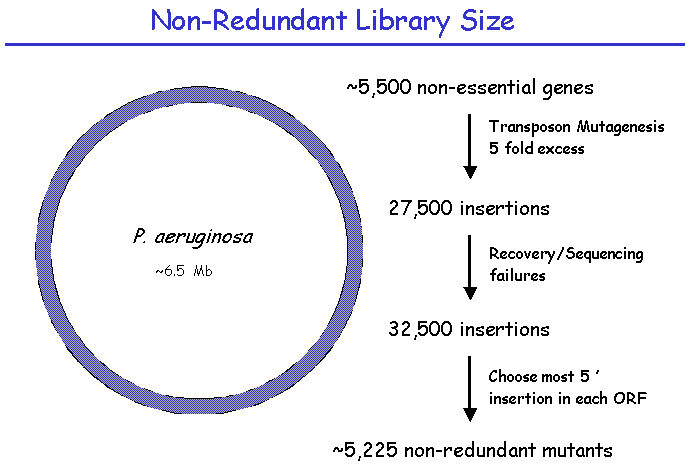
II. Library Construction: Library SizeThe construction of this library involves the production of approximately 30,000 random transposon insertions in the PA14 genome. This number gives a ~95% probability of obtaining an insertion in each of the non-essential PA14 genes. From this library of 30,000 sequenced insertions, a single insertion in each targeted gene is selected for inclusion in a PA14 “non-redundant” library. There are an estimated 5500 non-essential genes in PA14, which determines the size of the non-redundant library. This smaller library accelerates phenotype screening for a wide variety of phenotypes including both virulence and non-virulence-related functions. The larger 30,000 library provides alternative mutations in most genes, allowing confirmation of a gene's involvement in a phenotype of interest.
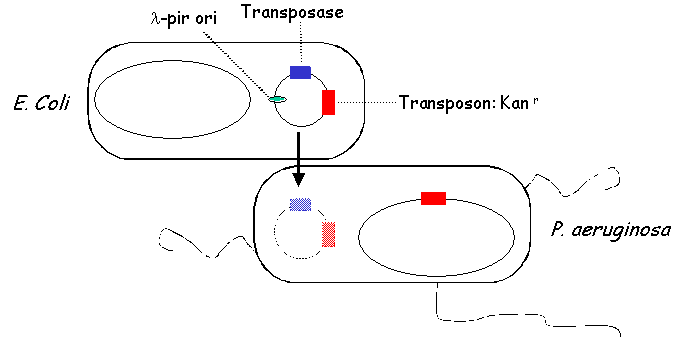
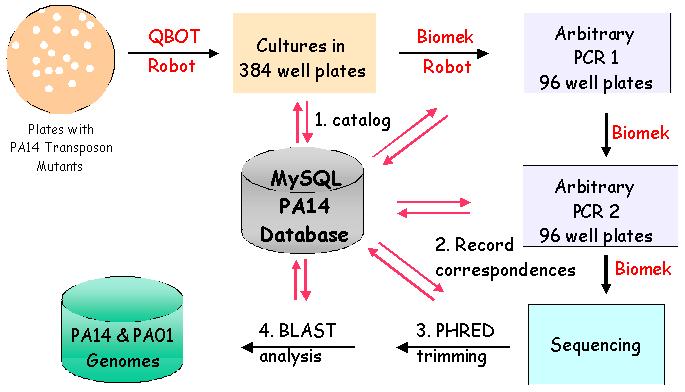
II. Library Construction: Generation of MutantsMutants are produced in the following way. E. coli carrying a plasmid that contains a tranposon, a transposase, a lambda-pir-dependent origin of replication and an antibiotic selection marker are mated with wild type PA14. Because PA14 is lambda pir-, the transferred transposon plasmid is not replicated in PA14. PA14 transposants are selected on media containing antibiotics that select for the presence of the transposon and that select against E. coli.
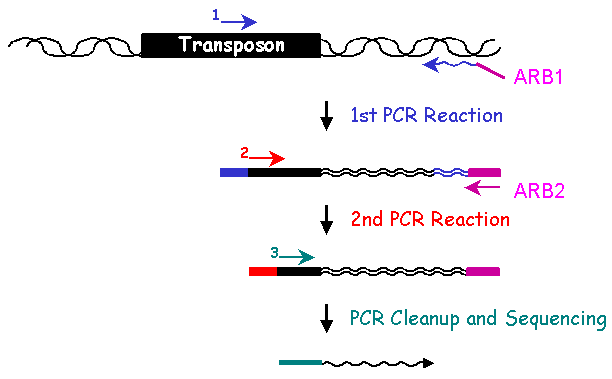
II. Library Constrution: Identification of Mutated GeneThe genomic DNA sequence adjacent to the transposon in each insertion mutant identifies the gene disrupted by the transposon. To determine the sequence of the adjacent DNA, two rounds of PCR are conducted. In the first round, a 5' primer specific to the transposon and a 3' ARB1 primer which contains a stretch of random nucleotides and a stretch of invariable nucleotides, are used to amplify genomic sequences. To enrich for genomic sequences adjacent to the transposon, a second round of PCR is performed using a nested 5' primer specific to the transposon sequence and a 3' ARB2 primer that anneals specifically with the invariant sequence present in the ARB1 primer. After cleaning up the ARB2 PCR reaction, another transposon-specific primer is used to sequence the Arbitrary PCR products. Subsequent bioinformatic analysis of the resulting sequences identifies the genomic locus adjacent to the transposon insertion.
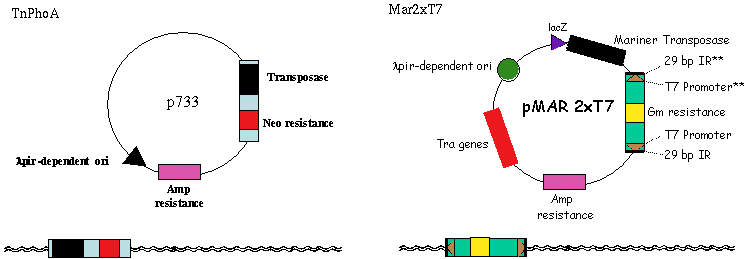
III. Transposons Used - TnPhoA and MarinerThe initial set of PA14 transposon mutants generated in this project were created using the Tn5-derived bacterial transposon, TnphoA. Although TnphoA exhibits relatively little site selectivity, we have identified at least one "hot spot" for Tn5 insertion in the PA14 gacA gene. Similarly, there are also most likely cold spots for Tn5 insertion in the PA14 genome. To avoid transposon site selectivity, we are also using the eukaryotic mariner transposon that has been shown to transpose in a variety of prokaryotes (77) and which presumably exhibits target site preferences different from that of TnphoA. John Mekalanos' laboratory at HMS has generated a series of mariner-based constructs for transposon mutagenesis in bacteria and has generously provided them for our studies in P. aeruginosa. Using these constructs we have demonstrated that the mariner transposon inserts into the PA14 genome very efficiently.
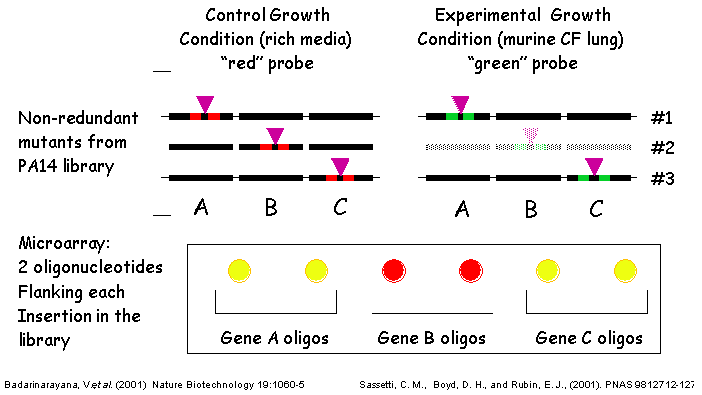
III. Transposons Used - TraSHTransposon site hybridization (TraSH) analysis is a high throughput method of screening for the presence of specific transposon mutants within bacterial pools subjected to different experimental conditions (Badarinarayana, V. et al. (2001) Nature Biotechnology 19:1060-5, Sassetti, C. M., Boyd, D. H., Rubin, E. J. (2001) PNAS 98:12712-12717) similar conceptually to the more commonly used Signature Tag Mutagenesis (STM) (Strauss, E. J., and Falkow, S (1997) Science 267: 707-12). Both screening strategies require that each strain contain a unique molecular tag such that the presence of each mutant can be determined within a complex pool. A group of strains can then be subjected to the experimental condition of interest, and all bacteria that are capable of surviving the treatment are isolated at the end of the experiment. Mutants in genes required for surviving under the experimental condition are those whose corresponding tags are underrepresented in the post-treatment population. In TraSH, the molecular tags used are the genomic DNA sequences directly adjacent to each transposon insertion. By engineering a T7 promoter at each end of the transposon, an in vitro transcription reaction from genomic DNA prepared from mutant pools will specifically amplify all of the tags present. These mRNAs are then reverse transcribed and fluorescently labeled to generate cDNA probes, which can then be hybridized to a P. aeruginosa microarray containing oligonucleotides complementary to these probes. In this manner, the relative fluorescence intensity for a particular oligonucleotide spot on the microarray corresponds to the relative number of bacteria present containing that tag. TraSH has been used successfully by the laboratories of George Church and Eric Rubin at Harvard Medical School to characterize growth preferences of E. coli and M. tuberculosis respectively (Badarinarayana, V. et al. (2001) Nature Biotechnology 19:1060-5, Sassetti, C. M., Boyd, D. H., Rubin, E. J. (2001) PNAS 98:12712-12717). To make the PA14 transposon mutant library compatible with future TraSH experiments, we have created a TraSH-compatible transposon construct, pMAR2xT7. To create pMAR2xT7, we have replaced extensive inverted repeat sequences at the ends of the mariner transposon in the Mekalanos lab mariner-based construct with the T7 promoter and short inverted repeat sequences present at the ends of the mariner transposon in pMycoMar, developed by C. Sassetti in the Rubin laboratory for TraSH analysis of Mycobacterium. We have confirmed that Mar2xT7 efficiently integrates into the PA14 genome (103 fold more frequently than TnphoA) resulting in single insertions, and RNA has been efficiently transcribed from the integrated T7 promoters. We plan to use pMAR2xT7 to create the majority of tranposon insertion mutants for the non-redundant library. TraSH analysis is dependent on a microarray chip to assess the presence or absence of mutants in a given mutant pool. The P. aeruginosa Spotted Oligonucleotide Array (described in PA14 Genomic Sequencing Project) will contain probes for all PAO1 and PA14 genes, as well as probes for additional P. aeruginosa strains. For each gene disrupted by a transposon insertion in our mutant library, TWO oligonucleotide probes will be designed, flanking the transposon insertion site to allow the same microarray to be utilized for TraSH analysis as described above.
IV. PA-TIMDB: Pseudomonas aeruginosa Transposon Insertion Mutant DatabaseWe have developed a corresponding relational database (PA14 Transposon Insertion Mutant Relational Database, or PA-TIMDB) to complement the non-redundant PA14 transposon insertion library. PA-TIMDB currently consists of three main parts: (1) a database to track information about the experimental status and location of each mutant sample, (2) automated sequence analysis for each sample to identify the locus of each mutation, and (3) a data-retrieval system to allow users to download data from PA-TIMDB over the web, including a list of mutants for which insertion locations have been identified. Mutants of interest from the library can be ordered through the on-line Mutant Request interface. V. Public Availability - Addition of Mutants to the DatabaseRather than offer only the completed non-redundant library, we are providing the public with mutants as we create them through PA-TIMDB. As of February 2004: Over 17,000 PA14 insertion mutants have been generated to date. A subset of these mutants have been processed by Arbitrary PCR and sequencing. Now that the PA14 sequence is complete, the rate of processing mutants and making them available to the public will increase rapidly. We plan to have the library nearly completed by October 2004. | |||||||||||||||||||||||||||||||||||||||